
Unraveling Biological Complexity With Charge Detection Mass Spectrometry
Characterizing the proteome
Protein-level biology holds the answers to some of the most pressing healthcare questions on researchers’ minds. From identifying important disease biomarkers to aiding drug development through mapping protein interactions, much can be gleaned from investigating the processes within the proteome. But how do we tap into this vast wealth of knowledge to continue advancing science?
Understanding the processes in the proteome requires characterization and analysis of the proteins contained. This is no easy task. The challenges around this protein analysis boil down to two main complicating factors: scale and complexity. First, the proteome can contain hundreds of proteins at different concentrations, necessitating techniques with large dynamic measurement ranges. Second, proteins exist not just as one individual species, but as a large number of proteoforms. These proteoforms are extremely difficult to characterize due to their subtle differences — yet this characterization is highly important, as proteoforms carry distinct biological functions that must be ascertained. Accurate and high-resolution analytical techniques are absolutely essential to decipher the complexity of the proteome, and to enable better biotherapeutic analysis and development.
A breakthrough in analyzing complex samples
The traditional tool of choice for characterizing large heterogeneous protein complexes is mass spectrometry (MS). MS measures the mass to charge ratio (m/z) of protein ions and offers high-resolution measurements that can address the complexity of proteoforms. Native MS, in particular, is an advantageous tool that can characterize the composition and stoichiometry of large heterogeneous protein complexes in their native state.
While a highly useful technique, there are still major challenges that exist with current MS approaches for large or complex macromolecule analysis. Most significantly, there can be an overlap in charge-state and isotopic distributions caused by residual solvation, ionic adducts and post-translational modifications. This overlap leads to a high level of complexity that is unresolved for larger complexes – around ~150 kDa – yielding spectra that are intractable to m/z-based deconvolution. Now, new developments in MS technology could hold the answer to analyzing previously intractable targets.
Finding clarity with Direct Mass Technology mode
Charge detection mass spectrometry (CDMS) is emerging as a solution to overcome the challenges in characterizing complex proteoforms by providing data directly in the mass domain for larger and more complex macromolecules. CDMS can be performed simultaneously with the traditional m/z measurements when using Thermo Scientific™ Orbitrap™ analyzer, equipping an existing well-established system with additional charge detection capabilities. This approach provides parallel individual ion measurements resulting in direct and accurate mass determination without requiring m/z-based deconvolution from charge-state or isotopically resolved signals in native ensemble measurements.
CDMS brings a host of advantages to large protein complex analysis. One of the most significant benefits is that resolution is increased up to 20-fold compared to traditional ensemble ion measurements. Plus, not only does it offer an increased dynamic range for better protein detection, but it also increases the upper limit of accessible mass measurements into the megadalton range. CDMS also requires less sample, or a less concentrated sample, due to its exquisite sensitivity to get excellent results compared to previous approaches.
So, how does it work? The sample is run, collecting hundreds of individual ions in parallel per spectrum (Figure 1). The m/z is determined by the frequency of axial oscillation, and Thermo Scientific™ Direct Mass Technology™ mode additionally calculates the integrated induced intensity signal over time, outputting a Selective Temporal Overview of Resonant Ions (STORI) plot. The slope of this plot of an individual ion is proportional to its charge, thereby allowing determination of z. From multiplying m/z and z, the mass of each individual ion can be calculated to give a high-resolution mass domain spectrum ready for analysis.
Figure 1: Schematic to show how high-resolution mass domain spectra are obtained via CDMS.
A tool that matches biological complexity
Researchers are exploring the potential of CDMS for the analysis of complex solutions of larger macromolecules. And the results are incredible. One study analyzed the potential of CDMS to measure proteoforms and their complexes in both denatured and native modes of operation. Over 500 proteoforms were detected by CDMS — standard ensemble MS alone detected none (Figure 2). Another study used CDMS to investigate intact proteoforms. Low-abundance fragment ions containing many hundreds of previously undetectable residues were identified, showing an increase of 48% in obtainable sequence coverage compared to the traditional readout for a 40 kDa protein. Without this technology, researchers would lack detailed insight and remain in the dark about the existence of any of these large species.
Complex biotherapeutic characterization can sometimes be achieved by traditional ensemble MS methods, but often requires multiple experiments or extensive sample processing. A recent study investigating Etanercept, a tumor necrosis factor (TNF) inhibitor, identified the coexistence of over 100 glycoforms using native MS and a Thermo Scientific™ Q Exactive™ Ultra-High Mass Range (UHMR) mass spectrometer. When using Direct Mass Technology mode instead of ensemble measurements, more confident characterization can be achieved to reveal the complex heterogeneity that exists in biotherapeutics with unit resolution (Figure 2).

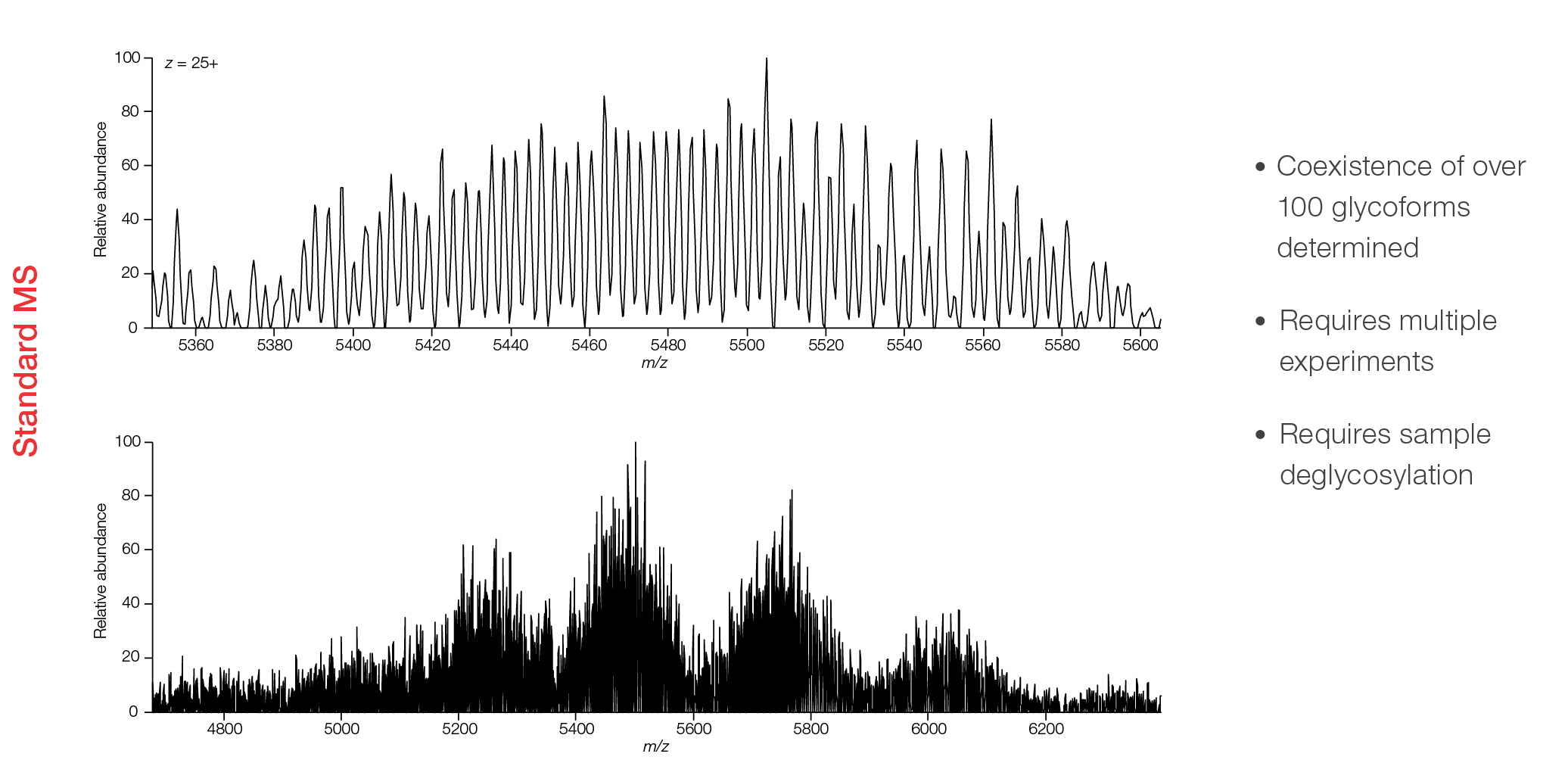
Figure 2: Analysis of the native Etanercept FC-fusion protein (approximately 125 kDa monomer) using a Q Exactive UHMR mass spectrometer with Direct Mass Technology mode (top), and traditional ensemble MS (bottom). Direct Mass Technology mode reveals a much wider proteoform distribution compared to the traditional approach.
CDMS is breaking records for the species size it can detect. In fact, CDMS has given unprecedented isotopic resolution for species with masses in the hundreds of kDa using an Orbitrap instrument with Direct Mass Technology mode. While plenty has been explored using CDMS, now that the technique is commercially available it has much to offer laboratories studying complex macromolecules.
Unlocking the potential of the proteome
Proteomes contain a vast wealth of information waiting to be drawn upon. CDMS gives us the ability to understand proteoforms in unprecedented detail and make the most of these valuable insights. Now, the immense complexity of biology can be explored in never-before-seen clarity, with even low frequency events observed. Not only is CDMS a vital tool for proteomic analysis, but its capabilities are ideal for application to complex biotherapeutic analysis, too. The ability to enhance our understanding of complex analytes will, ultimately, guide researchers and enable development of better biotherapeutics and next-generation drug modalities for improved healthcare.
About the author:
Daniel Hermanson began his research career in the field of chemical biology, focusing on utilizing mass spectrometry to understand biological processes. He earned his PhD from Vanderbilt University in Nashville, TN, studying the substrate-selective inhibition of cyclooxygenase-2. He then did post-doctoral research at the Scripps Research Institute applying Activity-Based Protein Profiling to study the function of unannotated proteins. Daniel joined Thermo Fisher Scientific as a field application scientist, supporting mass spectrometry customers with instrument and application training. He currently is a senior product marketing manager focused on advanced mass spectrometry products and software.






Gloss