b An input field allows the user to specify patterns of the total, bulk ligand concentrations. An association phase occurs during periods of non-zero bulk ligand concentration (e.g., 90–270 s for Ligand B). Dissociation phases occur when the ligand is removed from the bulk solution (e.g., 360–720 s for Ligand A). Here, Ligand C is specified as continuously present in solution during the 720 s of the interaction timecourse. The graphical display allows visualization of the specified bulk concentration pulse pattern. c User input parameters for the receptor. Receptor concentration can be specified as either an SPR-mimicking surface density (measured in RU; where 1 RU equals ~1 pg/mm2) or a molar concentration. Receptor topology is specified in the same form as described above for the ligands. d The MVsim controller tab enables initiation, iteration, and export of binding simulations. "Initiate" executes a simulation. "Re-run" executes an abbreviated simulation used when no changes were made to the valency or topology of the system. "Reset" relaunches the app and clears user input parameters from all fields. Credit: Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6">
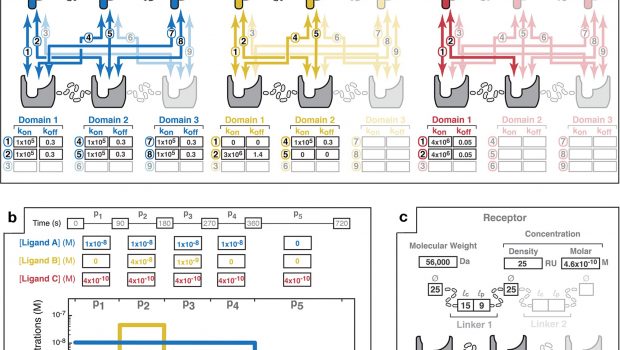
a A point-and-click interface enables the user to select the number of ligands (up to three) and valencies of the ligand(s) and receptor (up to trivalent) that compose the multivalent system. Based upon the chosen design, the user specifies the structure of each of the ligands by entering the applicable molecular weight (MW); the binding domain diameters (Ø); the contour lengths (lc of the linkers (i.e., the maximum end-to-end distance; e.g., 3.5 Å and 1.5 Å per amino acid for a random coil and alpha helix, respectively); and the persistence lengths (lp) of the linkers. Further, the applicable combinatorial interactions (numbered 1 to 9) unique to each receptor–ligand pairing are highlighted. Parameter fields allow the input of monovalent rate constants for each pairwise interaction. Non-binding interactions can be indicated with kon and koff values of zero (e.g., as illustrated with Ligand B in yellow for interactions "1" and "5"). b An input field allows the user to specify patterns of the total, bulk ligand concentrations. An association phase occurs during periods of non-zero bulk ligand concentration (e.g., 90–270 s for Ligand B). Dissociation phases occur when the ligand is removed from the bulk solution (e.g., 360–720 s for Ligand A). Here, Ligand C is specified as continuously present in solution during the 720 s of the interaction timecourse. The graphical display allows visualization of the specified bulk concentration pulse pattern. c User input parameters for the receptor. Receptor concentration can be specified as either an SPR-mimicking surface density (measured in RU; where 1 RU equals ~1 pg/mm2) or a molar concentration. Receptor topology is specified in the same form as described above for the ligands. d The MVsim controller tab enables initiation, iteration, and export of binding simulations. "Initiate" executes a simulation. "Re-run" executes an abbreviated simulation used when no changes were made to the valency or topology of the system. "Reset" relaunches the app and clears user input parameters from all fields. Credit: Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6" width="300"800" height="400"530"/>
The MVsim input design interface provides interactive parameter specification for systems of multivalent, multi-molecular interaction. a A point-and-click interface enables the user to select the number of ligands (up to three) and valencies of the ligand(s) and receptor (up to trivalent) that compose the multivalent system. Based upon the chosen design, the user specifies the structure of each of the ligands by entering the applicable molecular weight (MW); the binding domain diameters (Ø); the contour lengths (lc of the linkers (i.e., the maximum end-to-end distance; e.g., 3.5 Å and 1.5 Å per amino acid for a random coil and alpha helix, respectively); and the persistence lengths (lp) of the linkers. Further, the applicable combinatorial interactions (numbered 1 to 9) unique to each receptor–ligand pairing are highlighted. Parameter fields allow the input of monovalent rate constants for each pairwise interaction. Non-binding interactions can be indicated with kon and koff values of zero (e.g., as illustrated with Ligand B in yellow for interactions "1" and "5"). b An input field allows the user to specify patterns of the total, bulk ligand concentrations. An association phase occurs during periods of non-zero bulk ligand concentration (e.g., 90–270 s for Ligand B). Dissociation phases occur when the ligand is removed from the bulk solution (e.g., 360–720 s for Ligand A). Here, Ligand C is specified as continuously present in solution during the 720 s of the interaction timecourse. The graphical display allows visualization of the specified bulk concentration pulse pattern. c User input parameters for the receptor. Receptor concentration can be specified as either an SPR-mimicking surface density (measured in RU; where 1 RU equals ~1 pg/mm2) or a molar concentration. Receptor topology is specified in the same form as described above for the ligands. d The MVsim controller tab enables initiation, iteration, and export of binding simulations. "Initiate" executes a simulation. "Re-run" executes an abbreviated simulation used when no changes were made to the valency or topology of the system. "Reset" relaunches the app and clears user input parameters from all fields. Credit: Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6
A team led by University of Minnesota Twin Cities biomedical engineers has developed a universally accessible application that can simulate complex molecular interactions, which will allow researchers to design better treatments for diseases like cancer and COVID-19.
The paper builds upon a study the researchers published in 2019. Now, they've expanded the technology to simulate even more complex molecular interactions, made the application easy for non-experts to use, and applied their findings to shed light on how the SARS-CoV-2 virus infects the body.
The study is published in Nature Communications, and the app, called MVsim, is freely available to other researchers on GitHub.
The simulator predicts the strength, speed, and selectivity of multivalent interactions, which involve molecules that have multiple binding sites and can be used to develop medicines for diseases, particularly cancer and COVID-19.
"Multivalent interactions are really important in natural biological systems, and they are now starting to be creatively exploited for creating new therapeutic drugs that leverage their unique binding properties," said Casim Sarkar, senior author of the paper and a professor in the University of Minnesota Department of Biomedical Engineering.
"With multivalent drugs, you can, in principle, target cells very specifically in a way that's not possible with standard, monovalent drugs, but there are many variables to consider in their design and much of the work in the field to date has been done through experimental trial and error," Sarkar added. "Now, using MVsim, we're able to make good predictions that can be used to more rationally design such therapeutics."
Many cancer drugs not only bind to tumor cells but also to cells they aren't meant to target, which often creates unwanted side effects for the patient. By optimizing the specificity of multivalent interactions using MVsim, researchers can design drugs that more specifically target the cells in a tumor while minimizing binding to other cells in the body.
Another example is the SARS-CoV-2 virus. Scientists know that the virus is evolving to better infect our cells and evade our immune systems, but the molecular mechanisms behind how the virus does this is relatively unknown. Using their MVsim technology, the University of Minnesota researchers were able to explore this process more in depth, uncovering the rates at which individual binding domains within the virus's multivalent spike protein switch between a cell-infecting state and an immune-evading state.
"We essentially have a computational microscope that allows us to look under the hood and see what multivalent proteins such as the SARS-CoV-2 spike protein are doing at a molecular level," Sarkar explained. "This level of molecular detail is hard to capture with a physical experiment. One of the real powers of MVsim is that we can not only learn more about how these systems work but we can also use this tool to design new multivalent interactions for diseases like cancer and COVID-19."
The researchers have already identified potential ways to limit the infectivity of current and future SARS-CoV-2 variants, which they plan to test soon.
Engineered multivalent self-assembled binder protein against SARS-CoV-2 RBD
More information:
Bence Bruncsics et al, MVsim is a toolset for quantifying and designing multivalent interactions, Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6
Citation:
Technology that simulates complex molecular interactions could lead to better treatments for cancer and COVID-19 (2022, September 6)
retrieved 6 September 2022
from https://phys.org/news/2022-09-technology-simulates-complex-molecular-interactions.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.
Artica Proxy Unauthenticated PHP Deserialization… by Admin March 27, 2024 ### This module requires Metasploit: https://metasploit.com/download# Current source: https://github.com/rapid7/metasploit-framework##class MetasploitModule…(8)
We use cookies to ensure that we give you the best experience on our website. If you continue to use this site we will assume that you are happy with it.OkNoRead More
You can revoke your consent any time using the Revoke consent button.Revoke consent






Gloss